
FDA Zulassung von Medizinprodukten
Viele von Ihnen werden schon davon gehört haben, dass eine FDA Zulassung notwendig ist, um Medizinprodukte legal in die USA verkaufen zu können. Diese Verfahren sind beispielsweise eine Premarket Notification 510 (k) oder aber auch eine Premarket Approval (PMA). Es gibt auch ein weiteres, weniger bekanntes Verfahren. Das sogenannte De Novo Program der FDA. Aber „nur“ mit einer Produktzulassung ist es nicht getan. Es gibt neben den Zulassungen noch andere Anforderungen, die erfüllt werden müssen: Ihr Qualitätsmanagementsystem muss die Anforderungen der Quality Management System Regulation (QMSR) erfüllen und Produkte sowie Unternehmen müssen bei der Food and Drug Administration angemeldet werden. In diesem Blog erklären wir Ihnen, welche Schritte auf dem Weg zur US Zulassung notwendig sind.
FDA Zulassung bei der Food And Drug Administration (FDA)
Die FDA ist eine in den Vereinigten Staaten ansässige Behörde. Sie ist dem Gesundheitsministerium unterstellt und befasst sich mit der Überwachung von Lebensmitteln, Arzneimitteln und Medizinprodukten. Wie für eine derartige Behörde üblich, ist die Kernaufgabe der Schutz der Bürger. Um diese Aufgabe zu realisieren, hat dürfen bestimmte Produkte nicht einfach so in den USA verkauft werden. Sie brauchen eine FDA Zulassung.
Im Unterschied zu Behörden in Deutschland hat die FDA einen gewissen Einfluss auf die im Rahmen der FDA Zulassung einzuhaltenden Anforderungen. Sie hat die Befugnis bestimmte Gesetze zu erlassen, die dann im Rahmen der Zulassung zu beachten sind. Im Abschnitt 21 des Code of Federal Regulations (CFR) werden diese Gesetze aufgenommen.
Die FDA gilt weltweit als sehr strenge Behörde. Das führt dazu, dass sich die meisten Unternehmen vor einem FDA-Audit regelrecht fürchten. Natürlich hat die FDA die Möglichkeit, eine Einfuhrsperre oder ähnliche unangenehme Maßnahmen anzuordnen. Ein Verkaufsstop kann Ihnen aber auch in der EU passieren, wenn Anforderungen nicht eingehalten werden. Der Schlüssel zu einem ruhigen Schlaf ist also in der EU wie auch in den USA eine gute Vorbereitung und Umsetzung der regulatorischen Anforderungen.
Für viele Themen, die für die FDA Zulassung relevant sind, gibt es Guidelines. Diese geben den Erwartungshorizont der Behörde vor und helfen Ihnen tatsächlich dabei, sich regulatorisch gut aufzustellen.
Abweichungen & FDA Zulassung
Abweichungen im Rahmen der FDA Zulassung können während des Zulassungsverfahrens oder während der bestehenden Zulassung entstehen.
Während des Zulassungsverfahrens wird die FDA Ihnen die jeweiligen Abweichungen mitteilen und diese müssen durch Sie fristgerecht abgestellt werden. Die Fristen sind in den jeweiligen Verfahren vorgegeben. Da die Überprüfungen durch die FDA sehr detailliert sind, kommen Abweichungen in den Zulassungen häufig vor. Wichtig dabei ist, dass die Abweichungen nicht derart gravieren sind, dass es ein Problem im Projekt wird. Das schließen Sie aber durch eine gute Vorbereitung und Expertise in technischen und regulatorischen Aspekten weitestgehend aus. In unseren Projekten kam es bisher zu keinen derart schwerwiegenden Abweichungen, dass eine Zulassung gescheitert ist. Die während der FDA Zulassung auftretenden Abweichungen werden nicht veröffentlicht.
Abweichungen, die bei bestehender FDA Zulassung auftreten, also mit Produkten im Markt, können im Rahmen eines Warning letter veröffentlicht werden. Meldepflichtige Vorkommnisse werden in den USA über die FDA MAUDE Datenbank veröffentlicht.
Abweichungen werden üblicherweise in folgenden Stufen erfolgen:
- Stufe 1: FDA Form 483 (Abweichungsbericht durch die FDA)
- Stufe 2: Warning letter
- Stufe 3: Verkaufsstop
Als Beispiel: Wird eine Abweichung mittel Form 483 festgestellt und vom Unternehmen nicht ausreichend darauf reagiert, wird ein Warning Letter veröffentlicht. Wird wieder nicht ausreichend reagiert, wird die FDA einen Verkaufsstop aussprechen. Während des Prozesses hat der Hersteller die Möglichkeit, mit Vertretern der FDA über das Verfahren zu sprechen und Maßnahmen zu diskutieren. Ein Verkaufstop bricht also üblicherweise nicht unvorhergesehen über einen herein.
Durch die Veröffentlichung können Abweichungen zu Image-Schäden der Unternehmen führen. Sie können eine Liste veröffentlichter Warning Letter bei der FDA einsehen: Warning Letter.
*21 CFR Part 820 – QSMR
Bitte was?! Was sich hier etwas kryptisch anhört, ist die Referenz zur Quality Management System Regulation der FDA. Darin beschrieben sind die wichtigsten Anforderungen an ein Qualitätsmanagementsystem, mit dem Sie Produkte in den USA verkaufen dürfen. Hier wird auch beschrieben, welche Aufzeichnungen die FDA von Ihnen erwartet – Ein Blick in diese Anforderungen ist also schon lange vor einer US Zulassung interessant. Durch Umstellung von QSR auf QMSR werden die Anforderungen an das Qualitätsmanagementsystem stärker an die EN ISO 13485 angeglichen. In §820.7 „Incorporation by reference.“ Verweist die QMSR auf die ISO 9000 als Grundlage für diverse Begrifflichkeiten und auf die ISO 13485 hinsichtlich Anforderungen an das Qualitätsmanagementsystem. Diese Umstellung wird durch eine „Final Rule“ ausgelöst, die von der FDA herausgegeben wird und ab dem 02.02.2026 wirksam ist. An dieser Stelle erkennt man auch die weitreichenden Befugnisse der FDA.
Es ist absolut zu begrüßen, dass sich die FDA an den international bekannten und etablierten Anforderungen der ISO 13485 orientiert. Durch die Wirksamkeit ab 02.02.2026 ist es bis dahin trotzdem erforderlich, die bisherigen Anforderungen der QSR einzuhalten:
In 21 CFR 820 finden Sie Informationen zum Thema Design Controls bzw. Entwicklung von Medizinprodukten. An dieser Stelle empfiehlt es sich, die Anforderungen der QSR mit Ihrem bestehenden Entwicklungsprozess abzugleichen. Es mag sich simpel anhören, aber ein Hersteller muss Verfahren für die Definition und Dokumentation vom Design Output in einer Weise implementieren und aufrechterhalten, die eine angemessene Bewertung der Konformität mit den Anforderungen an den Design Input ermöglichen. Wir haben schon häufig festgestellt, dass Kunden nicht ganz klar war, was eigentlich der Design Output ihres Entwicklungsprozesses ist. In der Verifizierung wurden dann teilweise Aspekte geprüft, die nicht ausreichend belegen, dass die Anforderungen des Design Inputs im Design Output umgesetzt wurden.
Hinsichtlich der Anforderungen der Quality System Regulation sind Sie mit einem bestehenden QMS nach ISO 13485 schon gut aufgestellt. Doch es gibt viele Feinheiten, die es zu beachten gilt. Wir raten Ihnen dazu, mittels einer Gap Analyse herauszufinden, welche Anforderungen Sie noch nicht oder noch nicht ausreichend umgesetzt haben. Stellen Sie also vor einer US Zulassung sicher, dass Ihr Unternehmen auf Ebene des QMS gut vorbereitet ist.
Klassifizierung von Medizinprodukten zur FDA Zulassung
Die Verfahren zur FDA Zulassungen hängen im Grunde mit dem Produktrisiko zusammen. Je risikoreicher ein Produkt, desto höher ist also seine Risikoklasse. Wichtig zu beachten ist, dass die FDA eigene Klassifizierungen hat. Die Klassen aus der EU können nicht einfach übertragen werden. Das gilt im Übrigen auch dafür, was überhaupt als Medizinprodukt eingestuft wird oder eben nicht.
Die FDA kennt drei Risikoklassen:
| Klasse | Beschreibung |
|---|---|
| I | Niedriges Risiko: Zulassung über Listing oder 510(k) |
| II | Mittleres Risiko: Zulassung über Listing oder 510(k) |
| III | Hohes Risiko: Zulassung über PMA (nur selten über 510(k)) |
Verwendung der "Device Classification Panels" in der FDA Zulassung
Die meisten Medizinprodukte können dadurch klassifiziert werden, dass über die Medical Specialty des Device Classification Panels die passende Kategorie ausgewählt wird. Das zeigt die folgende Abbildung.
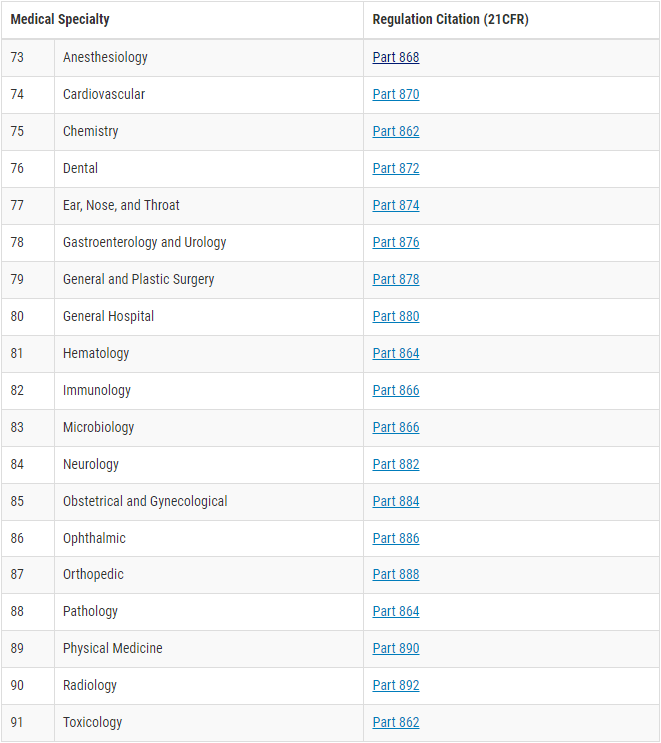
Nehmen wir an, wir möchten einen PCR Test zum Nachweis von HIV zulassen. Wir öffnen also die Medical Specialty 82 Immunology und suchen dort nach dem passenden Produkttyp.

Ein PCR Test ist ein Testverfahren, das auf der Nukleinsäure-Amplifikations-Technik (NAT) beruht. Der richtige Eintrag ist also „§ 866.3957 – Human immunodeficiency virus (HIV) nucleic acid (NAT) diagnostic and/or supplemental test.“
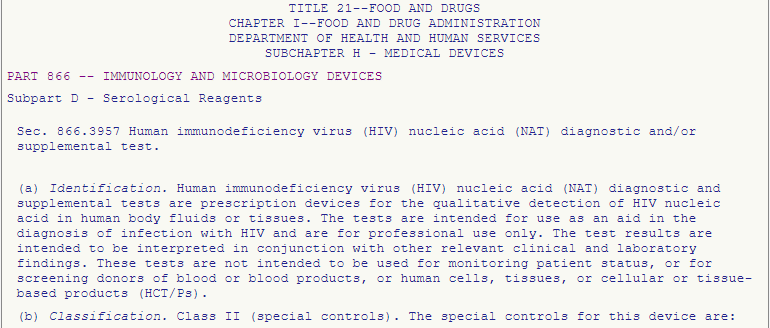
Das Produkt ist also in Klasse II eingestuft und muss die unter § 866.3957 aufgeführten Special Controls erfüllen.
Ermittlung eines geeigneten FDA Zulassungsverfahrens
Wir haben bereits beschrieben, dass Sie Ihre Produkte listen und Ihr Unternehmen registrieren müssen. Doch um Produkte listen zu können, müssen Sie diese zunächst einmal zulassen – sofern erforderlich. Ist dies nicht nötig, benötigen Sie für das Produkt auch keine eindeutige Registrierungsnummer, die Sie beim erfolgreichen Durchlaufen einer Zulassung bekommen. Sie können die Produkte also ohne Zulassung bei der FDA listen.
Doch gehen wir einmal davon aus, dass Ihr Produkt formal zugelassen werden muss und ein Listing nicht ausreicht. Woher wissen Sie, ob Sie eine Premarket Notifikation 510(k), eine Premarket Approval oder den Weg über das De Novo Program der FDA nutzen sollen?
Kennen Sie die Risikoklasse Ihres Produkts? Dies ist ein erster wichtiger Anhaltspunkt, um eines der Zulassungsverfahren zu wählen. Kennen Sie den Product Code, über den die passende Produktart bei der FDA reguliert wird? Das ist ideal! Denn darüber können Sie herausfinden, welche Anforderungen Sie einhalten müssen.
Ermittlung eines Product Codes zur FDA Zulassung
Product Codes können Sie mit Hilfe der 510(k) Premarket Notification Database oder der Premarket Approval (PMA) Database der FDA herausfinden. Geben Sie beispielsweise beim Feld Device Name der 510(k) Datenbank ein Produkt eines anderen Herstellers ein, das Ihrem Produkt ähnelt. Im vorliegenden Beispiel nutzen wir die ungefähre Bezeichnung eines Endoskops eines bekannten Herstellers. Wir haben keine Genehmigung für die Verwendung des Produktnamen und dazugehöriger Informationen eingeholt. Aus diesem Grund sind die entsprechenden Felder in den Grafiken gelöscht.

Nachdem Sie den ungefähren Device Name eingegeben haben, wird Ihnen eine Liste mit Suchergebnissen angezeigt. Wählen Sie das passendste Ergebnis aus. Anschließend wird Ihnen folgende Ansicht gezeigt, aus der Sie den gesuchten Product Code entnehmen können.
Wie Sie sehen, sind diesem Endoskop sogar mehrere Product Codes zugewiesen. Dies hängt mit der Zweckbestimmung zusammen und führt dazu, dass Sie die Anforderungen an jeden einzelnen Product Code einhalten müssen.

Sie können nun die Codes und die Regulation Number öffnen. Für den Code NWB wird Ihnen beispielsweise Folgendes angezeigt:

In dieser Übersicht wird Ihnen angezeigt, dass Sie eine 510 k als Zulassungsart nutzen müssen. Die recognized consensus standards sind Normen, die Sie für Produkte dieses Codes einhalten sollten. Aus der Regulation Number entnehmen Sie die gesetzliche Grundlage aus dem Code of Federal Regulations (21 CFR).
Auf diese Weise können Sie Schritt für Schritt die Anforderungen an jeden einzelnen Product Code ermitteln, der für Ihr Produkt anwendbar ist. Im „Idealfall“ ist es natürlich nur einer, das macht Ihnen das Leben am einfachsten. Aber wie Sie sehen gibt es Produkte, bei denen die Zweckbestimmung und Funktionsumfang dazu führen, dass es mehrere Codes werden.
FDA Zulassung: Zulassungsarten gemäß Product Code
- Für Klasse III Produkte wird in der Regel eine Premarket Approval (PMA) benötigt.
- Sofern Sie einen Product Code ermitteln konnten, ein Predicate Device verfügbar ist und die Product Codes eine 510(k) erfordern, können Sie den Weg über eine Premarket Notification gehen, um Ihr Produkt zuzulassen. Der Vergleich mit dem Predicate Device ist dabei die Argumentationsgrundlage in der Zulassung. Alle Unterschiede werden diskutiert und auf ihren Einfluss hinsichtlich der Sicherheit und Leistung beurteilt.
- Sie konnten keinen passenden Product Code zu Ihrem Produkt ermitteln? Ihre Produktart wurde somit noch nicht bei der FDA zugelassen. Neue Produkte wie dieses werden bei der FDA zunächst in Klasse III eingestuft. Wenn Sie Ihr Produkt in Klasse I oder II einstufen lassen möchten, ist das De Novo Program der FDA die richtige Methode.
- Ihr Produkt erfordert keine FDA Zulassung? In diesem Fall müssen Sie das Produkt dennoch bei der FDA listen.
Registrierung auch ohne FDA Zulassung: „Registration and Listing“
Unternehmen, die an der Herstellung und dem Vertrieb von Medizinprodukten zur Verwendung in den Vereinigten Staaten beteiligt sind, müssen sich jährlich bei der FDA registrieren lassen. Dieser Prozess wird als „establishment registration“ bezeichnet. Im Jahr 2020 beliefen sich die Kosten dafür auf 5.236 USD. Für das Fiskaljahr 2026, welches am 01.10.2025 beginnt, belaufen sich die Kosten auf 11.423 USD.
Die meisten Unternehmen, die sich bei der FDA registrieren lassen müssen, müssen auch ihre Produkte für den US Markt listen. Wenn ein Produkt vor dem Inverkehrbringen in den USA eine Premarket Approval oder Premarket Notification benötigt, ist die Identifikationsnummer der Zulassung mit anzugeben. Die Registrierung und das Listen erfolgen elektronisch.
U.S. Agent für die FDA Zulassung
Wenn Ihr Unternehmen als „foreign establishment“ gilt und an der Herstellung, Vorbereitung, Verbreitung, Zusammenstellung oder Verarbeitung eines in die USA eingeführten Produkts beteiligt ist, benötigen Sie einen U.S. Agent.
Der U.S.-Agent muss entweder in den USA wohnhaft sein oder dort eine Niederlassung unterhalten. Seine Verantwortlichkeiten beziehen sich auf:
- die Unterstützung der FDA bei der Kommunikation mit Ihrem Unternehmen.
- die Beantwortung von Fragen bezüglich Ihrer Produkte, die Sie in den USA in Verkehr bringen wollen.
- die Unterstützung der FDA bei der Planung von Inspektionen in Ihrem Unternehmen.
- Wenn die FDA nicht in der Lage ist, Ihr Unternehmen direkt oder schnell zu kontaktieren, kann die FDA dem U.S. Agent Informationen oder Dokumente zur Verfügung stellen, und eine solche Handlung wird als gleichwertig mit der Bereitstellung derselben Informationen oder Dokumente an Ihr Unternehmen angesehen.
Im Rahmen Ihrer „facility registration“ geben Sie den U.S. Agent namentlich an. Dies geschieht elektronisch über das FDA Unified Registration and Listing System (FURLS-System).
FDA Zulassung: wichtige Verfahren im Überblick
FDA Pre-Submission-Programm
Beim sogenannten Pre-Sub der FDA handelt es sich um ein formales Verfahren, das noch vor der eigentlichen Zulassung steht. Es dient dazu, offene Fragen zur Strategie zu klären und kommt in der Regel vor Verfahren wie 510(k), De Novo Request oder PMA.
Premarket Notification (PMN) oder 510(k)-Zulassung
Das 510(k)-Zulassungsverfahren ist nicht für neuartige Produkte möglich, sondern kommt dann zum Zug, wenn es bereits vergleichbare zugelassene Produkte gibt. Bei einer 510(k) wird sich zur Zulassung auf ein bereits zugelassenes Produkt bezogen, was insbesondere hinsichtlich der Notwendigkeit klinischer Daten hilfreich ist. Dadurch ist ein vergleichsweise schneller Marktzugang möglich und die 510(k) ist als das wichtigste Verfahren für die FDA Zulassung von Medizinprodukten in den USA anzusehen.
Für diesen Prozess stehen mehrere Optionen zur Verfügung. Die traditionelle 510(k)-Variante ist für Produkte mit ähnlichen Vorgängerprodukte vorgesehen, wohingegen die Special 510(k)-Variante für Änderungen eigener Produkte genutzt wird, wozu wiederum ein beschleunigtes Verfahren eingesetzt wird. Schließlich gibt es noch das sogenannte Abbreviated 510(k)- Verfahren, das die Normenkonformität mit den „Special Controls“ und „Guidance Documents“ prüft. Mittlerweile bietet das eStar-Program eine Möglichkeit zur digitalen FDA Zulassung von 510(k)s
Premarket Approval (PMA)
Neuartige Produkte oder Produkte der Klasse III müssen das PMA-Zulassungsverfahren durchlaufen, was sich typischerweise ziemlich aufwendig sowie zeitintensiv gestaltet und sich daher für Hersteller von eigentlich unkritischen Produkten als zu aufwändig erweist. Der Grund für die erhöhten Anforderungen: Zur Klasse III zählen Hochrisikoprodukte, die entsprechend hohe Anforderungen an ihre Zulassung haben.
De Novo Classification Request / De Novo Verfahren
Dabei handelt es sich um das FDA Zulassungsverfahren für neuartige Medizinprodukte. Das bedeutet, dass es um ein Produkt geht, für welches es kein bei der FDA zugelassenes Produkt (Predicate Device) gibt, das aber in Klasse I oder II eingeteilt werden soll. Dabei unterscheidet man zwischen dem klassischen de novo Verfahrensansatz und dem direct de novo Ansatz:
Der klassische Ansatz beginnt mit dem 510(k)-Antrag, der Premarket Notification. Wird dieser von der FDA abgelehnt, weil es sich um ein neues Produkt handeln könnte, kann der Hersteller den direct de novo Antrag stellen, was sich allerdings nur dann als zielführend erweist, wenn der Hersteller davon ausgehen darf, dass kein vergleichbares Produkt existiert und das eigene Produkt vermutlich nach Klasse I oder II eingestuft werden wird.
FDA Zulassung: Zusammenfassung
Schon vor der Produktzulassung in den USA gibt es vieles zu beachten:
- Ihr Qualitätsmanagementsystem muss die Anforderungen der Quality System Regulation 21 CFR Part 820 und ggf. weitere regulatorische Anforderungen erfüllen.
- Es ist zu beachten, dass eine Umstellung auf die Quality Management System Regulation erfolgt und somit eine Annäherung an die ISO 13485.
- Sie müssen Ihr Unternehmen bei der FDA anmelden und einen U.S. Agent benennen.
- Anschließend gilt es, unter Berücksichtigung der Risikoklasse und des Product Codes, das richtige Zulassungsverfahren und die für das Produkt anwendbaren Anforderungen, Normen und Guidances zu identifizieren.
- Erst dann beginnt die Produktzulassung über eine Premarket Notification 510(k), Premarket Approval oder als De Novo.
Professionelle Hilfe bei der FDA Zulassung für Medizinprodukte
Wenn Sie Hilfe bei der FDA Zulassung benötigen, sind wir von thinqbetter in jedem Fall der richtige Partner für Sie.
- Unsere Qualifikation – Durch die Betreuung von mehr als 140 unterschiedlichen Unternehmen können wir umfangreiche Erfahrungen in diversen Fachgebieten in die Waagschale werfen. Darüber hinaus arbeiten wir stetig an der Optimierung und Verbesserung unserer Arbeitsprozesse, Projektlaufzeiten und Qualität. Unser Team aus Fachkräften verschiedener Naturwissenschaften (z.B. Medizintechnik, Biologie, Chemie, Biotechnologie, Immunologie) wird überdies regelmäßig geschult, um für alle angebotenen Dienstleistungen zu jedem Zeitpunkt optimal gewappnet zu sein.
- Unsere Werte – Wir handeln verlässlich sowie seriös und genießen das Vertrauen vieler langjähriger Kunden. Die Medizintechnik ist streng reguliert und die Vielzahl unterschiedlicher Anforderungen stellt für Hersteller eine große Herausforderung dar. Deshalb stehen wir Ihnen als zuverlässiger, kompetenter und vertrauensvoller Partner jederzeit zur Seite – sowohl als Berater als auch als Dienstleister. Wir setzen uns dafür ein, dass Ihre Produkte zuverlässig die FDA Zulassung
- Unser Leistungsumfang im Bereich FDA Zulassung – Wir haben unsere Angebote im Bereich B2B für Hersteller von Medizinprodukten und Hersteller von In-vitro-Diagnostika (IVDs) maßgeschneidert. Neben einer kostenlosen Erstberatung können wir Ihnen im Bereich der FDA Zulassung folgende Leistungen anbieten:
- Erstellung bzw. Zusammenstellung oder Unterstützung bei der Erstellung von Premarket Notifications, auch Premarket Notifications 510(k) / 510(k) / PMN / PMN 510(k) genannt.
- Erstellung / Zusammenstellung oder Unterstützung bei der Erstellung von Premarket Approvals
- Erstellung / Zusammenstellung oder Unterstützung bei der Erstellung von De Novo Classification Requests
- Erstellung / Zusammenstellung oder Unterstützung bei der Erstellung von Presubmissions vor einer FDA Zulassung
- Unterstützung bei Facility Registration und Device Listing bei der FDA
Hinzu kommen weitere Dienstleistungen sowie Beratungen zu regulatorischen Themen.
Wenn Sie Unterstützung bei der bevorstehenden FDA Zulassung benötigen, nehmen Sie am besten gleich Kontakt zu uns auf. Das gibt uns ausreichend Zeit, alle notwendigen Maßnahmen zu ergreifen und Sie sicher durch die FDA Zulassung zu führen!
Häufig gestellte Fragen
Welche Bedeutung hat der Warning Letter bei der FDA Zulassung?
Wird eine signifikante Verletzung der Anforderungen bei bestehender FDA Zulassung festgestellt, kann die FDA den Hersteller darüber im Rahmen eines Warning Letter formal informieren. Dabei kann es sich beispielsweise um die Nichterfüllung notwendiger Anforderungen an Produktionsprozesse oder unzulässige Werbeaussagen handeln. In diesem Fall sind umgehend entsprechende Korrekturmaßnahmen einzuleiten. Innerhalb von 15 Tagen muss daher ein Korrekturplan vorgelegt werden. Nichtreaktionen könnten schlimmstenfalls zu einer strafrechtlichen Verfolgung führen. Ein Warning Letter wird von der FDA veröffentlicht und ist für jeden einsehbar.
Welche Voraussetzungen sind für eine FDA Zulassung wichtig?
Der Begriff „Zulassung“ wird meist für mehrere Dinge genutzt, obwohl es sich dabei nicht immer um dasselbe handelt. Der Begriff FDA Zulassung wird meist als Synonym dafür verwendet, dass ein Medizinprodukt ein beliebiges Verfahren durchlaufen hat, wodurch es in den USA verkauft werden darf. Die FDA selbst spricht bei einer 510(k) von einer sogenannten „Clearance“. Auf Begriffe wie „FDA Approval“ sollte verzichtet werden. In jedem Fall müssen die jeweiligen gesetzlichen Vorgaben an das Produkt und das Qualitätsmanagementsystem erfüllt werden.
Welche Unterschiede gibt es zwischen der FDA Zulassung und der EU-Zulassung?
Sowohl die USA als auch die EU wollen die Konformität von Medizinprodukten sicherstellen. Obwohl die Ziele praktisch gleich sind, gibt es beispielsweise Unterschiede zwischen den Produktklassifizierungen und Zulassungsverfahren. Einer der Hauptunterschiede besteht in der Möglichkeit, eine Premarket Notification 510(k) ohne klinische Daten durchzuführen, auch wenn in der EU beispielsweise eine klinische Prüfung nötig wäre. Ein weiterer Unterschied liegt in dezentralen Zulassungsfahren durch benannte Stellen gegenüber der zentralen, behördlichen FDA Zulassung.