- Medizinprodukte Klasse 1
- 22. Dez. 2022
Zulassung von Medizinprodukten der Klasse 1
Lukas Losigkeit
CEO & Principal Consultant
- Was sind Medizinprodukte?
- Was bedeutet Klasse 1?
- Welche Medizinprodukte sind in Klasse 1 (Beispiele)?
- Welche Fristen gibt es?
- Was bedeutet es, Hersteller zu sein?
- Auslagerung der Produktion und Beschaffung von Rohstoffen
- Wie erfüllt man die MDR?
- Qualitätsmanagement-System
- Technische Dokumentation
- Erstellung einer EU-Konformitätserklärung
- Anmelden in DMIDS
- Aufrechterhaltung der technischen Dokumentation
- Lokale Aufsichtsbehörde & Inspektionen - Hilfe! Was jetzt?
- Streitfall Klassifizierung: Mein Produkt ist Klasse 1 - die lokale Behörde sieht das anders.
Was sind Medizinprodukte?
Einfach gesagt sind Medizinprodukte Produkte, die einen medizinischen Zweck erfüllen und maßgeblich physikalisch wirken, um ihre Leistung zu entfalten. Um wirklich genau festzulegen, ob es sich im vorliegenden Fall um ein Medizinprodukt handelt oder nicht, muss anhand der Definition überprüft werden:
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben
...und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung.
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
Was bedeutet Klasse 1?
Medizinprodukte werden in die Klassen I, IIa, IIb, und III eingestuft. Die niedrigste Klasse (1 bzw I) ist für Produkte mit niedrigem Risiko für Patienten. Je höher die Risikoklasse, desto höher ist das zugrundegelegte Risiko des jeweiligen Medizinprodukts.
Zugegeben, einige Medizinprodukte werden unter der MDR in Risikoklassen eingestuft, die vielleicht etwas übertrieben erscheinen. Daran lässt sich allerdings nichts ändern - Es sind strikt alle Klassifizierungsregeln der MDR zu berücksichtigen. Die jeweils höchste Risikoklasse, die aus der Anwendung aller Klassifizierungsregeln resultiert, gibt also die tatsächliche Risikoklasse eines Produkts vor.
Welche Medizinprodukte sind in Klasse 1 (Beispiele)?
- Rollatoren und viele andere Hilfsmittel
- Pflaster und Kompressen
- Physiotherapieliegen
- Wiederverwendbare chirurgische Instrumente
- usw.

Welche Fristen gibt es?
Medizinprodukte der Klasse 1 müssen zu den folgenden Fristen die Anforderungen der MDR erfüllen:
- Medizinprodukte, die unter MDD (93/42/EWG) in Klasse 1 waren und unter MDR (EU 2017/745) auch in Klasse 1 sind, müssen seit dem 26.05.2021 die MDR erfüllen.
- Medizinprodukte, die unter MDD (93/42/EWG) in Klasse 1 waren und unter MDR (EU 2017/745) in eine höhere Klasse eingestuft werden, haben längere Übergangsfristen. Dadurch soll dem stark erhöhten Aufwand durch die Einbeziehung einer benannten Stelle Rechnung getragen werden. Ein Inverkehrbringen ist bis 26.05.2024 gestattet.
- Medizinprodukte, die unter MDD (93/42/EWG) in Klasse 1r, 1m, 1s, IIa, IIb, III gefallen sind, haben wiederum andere Fristen. Auf diese gehen wir im vorliegenden Artikel jedoch nicht ein.
Hinweis: Sollte in Ihrem Fall eine andere Frist als 26.05.2021 gelten, so muss Ihr Qualitätsmanagement-System die gesetzlichen Mindestanforderungen trotzdem seit dem 26.05.2021 erfüllen!
Was bedeutet es, Hersteller zu sein?
Beispiel für ein Medizinprodukt Klasse I
Es gibt unterschiedliche Wege, wie die Rolle eines Herstellers von Medizinprodukten genau aussehen kann. Dennoch haben alle eines gemeinsam: Das Unternehmen, das im rechtlichen Sinne als Hersteller eines Medizinprodukts auftritt, hat alle Rechte und Pflichten eines Herstellers. Dazu ist insbesondere Artikel 10 der MDR (EU 2017/745) zu den allgemeinen Pflichten der Hersteller zu beachten. Es gibt allerdings auch Aspekte der Produkthaftung, die relevant sind.
Szenario 1 - Ich stelle selbst Medizinprodukte her: Die Produktion von Medizinprodukten findet im eigenen Unternehmen statt. Dementsprechend habe ich selbst die volle Verantwortung für das, was in der Produktion geschieht.
Szenario 2 - Ich stelle selbst keine Medizinprodukte her: Die Produktion von Medizinprodukten wird von einem Unterauftragnehmer durchgeführt. Im Rahmen seiner vertraglichen Verpflichtung ist dieser somit für die von ihm erbrachten Leistungen verantwortlich. Wichtig dabei zu beachten ist allerdings, dass Sie als Hersteller im rechtlichen Sinne dennoch die finale Verantwortung für das haben, was in der Produktion geschieht. Sie sind es, der die Medizinprodukte in Verkehr bringt und Sie haben die Verantwortung, dass diese Produkte MDR-konform sind. Sie lagern also lediglich die Produktion aus, aber niemals die Verantwortung!
Szenario 3 - Mischform aus Szenario 1 und 2: Einige Hersteller haben ein breiteres Portfolio und produzieren intern und mit Unterauftragnehmern. Selbstverständlich sind Sie auch hier für alle durch Sie in Verkehr gebrachten Produkte vollständig verantwortlich.
Auslagerung der Produktion und Beschaffung von Rohstoffen
Es kommt oft vor, dass Hersteller die Produktion und gleichzeitig die Beschaffung der dafür notwendigen Rohstoffe an einen Unterauftragnehmer ausgelagert haben. Leider kommt es dabei oft auch vor, dass die Verantwortung für die Produktqualität und -Konformität beim Unterauftragnehmer vermutet wird. Das ist allerdings nicht zulässig.
Sie können den Prozess der Produktion und Beschaffung von Rohstoffen natürlich auslagern. Wie in den Szenarien 1 bis 3 bereits beschrieben, bleibt die Verantwortung allerdings bei Ihnen. Sie müssen in jedem Fall die internen und ausgelagerten Prozesse ausreichend überwachen, um die Einhaltung der regulatorischen Anforderungen sicherzustellen.
Im Falle der ausgelagerten Produktion bedeutet das, dass Sie die Herstellungsprozesse überprüfen und freigeben müssen. Es darf nur nach den vereinbarten Abläufen produziert werden. Und Sie müssen die Einhaltung dessen beispielsweise durch Audits und Chargenfreigaben überprüfen. In jedem Fall sollten Sie eine QSV (Qualitätssicherungsvereinbarung) schließen. Also einen Vertrag mit dem Unterauftragnehmer, in dem die jeweiligen Rechte und Pflichten beider Seiten nachvollziehbar geregelt sind. So ein Vertrag schützt Sie auch im Falle von Fehlern des Unterauftragnehmers. Es ist also wirklich in Ihrem Interesse, QSVs abzuschließen. Ein Rückruf führt schnell zu Kosten im 6-stelligen Bereich.
Wenn ein Unterauftragnehmer die Beschaffung von Rohmaterial zur Produktion übernimmt, müssen Sie auch die dafür notwendigen Wareneingangsprüfungen und sonstigen In-Prozess-Kontrollen vereinbaren. Wie gesagt: Sie lagern die Beschaffung aus, nicht die Verantwortung.
Beachten Sie folgende Faustformel:
Ausgelagerte Prozesse müssen Sie mindestens in gleichem Maße überwachen und kontrollieren wie interne Prozesse. Sie lagern Prozesse aus, nicht die Verantwortung des Herstellers von Medizinprodukten!
Wie erfüllt man die MDR?
Grundsätzlich gibt Artikel 10 der MDR mit dem Titel "Allgemeine Pflichten der Hersteller" einen guten Überblick, was getan werden muss. Das Ganze leichter verdaulich, stellen wir im folgenden Text dar. Grundsätzlich sollten Sie jedoch beachten, dass alle der nachfolgenden Punkte zu erfüllen sind. Es reicht für einen legalen Zustand nicht aus, wenn nur einige der Punkte erfüllt werden.
Qualitätsmanagement-System
Technische Dokumentation
- Jedes Produkt benötigt eine klinische Bewertung.
Dazu gibt es hier nähere Informationen. - Jedes Produkt benötigt eine Risikomanagement-Akte.
Dazu gibt es hier nähere Informationen. - Jedes Produkt mit patientenkontakt muss hinsichtlich seiner Biokompatibilität beurteilt werden.
Dazu gibt es hier nähere Informationen. Erstellung einer EU-Konformitätserklärung
Anmelden in DMIDS
Aufrechterhaltung der technischen Dokumentation
Als Hersteller, der ausschließlich Medizinprodukte der Klasse 1 im Portfolio hat, benötigen ein Qualitätsmanagement-System, das die gesetzlichen Mindestanforderungen erfüllt. Ganz wichtig dabei ist es, dass so ein QM-System nicht nach ISO 9001 oder ISO 13485 zertifiziert sein muss - aber kann!
Insbesondere für kleine Hersteller und Start-Ups, die gerade mit einem Produkt der Klasse 1 den Weg in die Medizintechnik wagen, ist dieser Hinweis sehr wichtig. Die Implementierung und Aufrechterhaltung eines zertifizierten QM-Systems ist wesentlich aufwändiger, als lediglich die gesetzlichen Mindestanforderungen abzudecken.
Aber auch wenn Sie den weniger aufwändigen Weg gehen möchten bzw. können - Bedenken Sie, dass der Betrieb eines QM-Systems parallel zum Arbeitsalltag stattfindet. Das bedeutet, planen Sie einen gewissen Aufwand dafür ein. QM ist nichts, was man am Jahresende nachdokumentiert und dann auf der sicheren Seite ist.
Ein funktionierendes QM-System ist die grundlage dafür, dass Ihr Geschäftsmodell dauerhaft legal und lebensfähig ist!
Ein QM-System nach gesetzlichen Mindestinhalten ist ein System, das Sie dazu in die Lage versetzt, in der Medizintechnik und der Europäischen Union mitzuspielen. Sie haben also verschriftlichte Prozesse, die nachvollziehbare Vorgaben dazu machen, wie Sie beispielsweise mit Reklamationen umzugehen haben. Denn jede Reklamation muss von Ihnen als Hersteller dahingehend überprüft werden, ob es sich um ein sogenanntes "schwerwiegendes Vorkommnis" handelt. Dieses müssten Sie dann an Ihre lokale Aufsichtsbehörde melden.
Wenn Sie nicht wissen, was so ein "schwerwiegendes Vorkommnis" ist: Auch das ist in Prozessen nachvollziehbar beschrieben. Sie werden also mittels QM-System dazu in die Lage versetzt, sich richtig als Hersteller von Medizinprodukten zu verhalten. Sie sind dazu in der Lage, auf Ereignisse rechtskonform zu reagieren und können so den gesetzlich nötigen Beitrag zur Sicherheit von Personen und insbesondere Patienten leisten.
Prinziell können Sie sich das QM-System also als die Befähigung eines Unternehmens vorstellen, sich wie ein Hersteller von Medizinprodukten zu verhalten.
Die technische Dokumentation wird je Produkt erstellt. Das ist im Grunde eine Produktakte, in der Nachweise darüber enthalten sein müssen, dass das Produkt die gesetzlichen Anforderungen erfüllt.
Hinweis: Die Zusammenfassung von technischen Dokumentationen sind der Einfachheit halber nicht beschrieben.
Die gesetzlichen Anforderungen an Produkte werden dabei maßgeblich in den sogenannten "Grundlegenden Sicherheits- und Leistungsanforderungen" in Anhang I der MDR beschrieben. Es ist also je Produkt zu überprüfen, welche dieser Anforderungen anwendbar sind. Die Einhaltung aller anwendbaren Anforderungen muss nachgewiesen werden. Das bedeutet: Es sind Dokumente dafür notwendig. Dinge wie "das war schon immer so" oder "das Produkt ist seit 20 Jahren auf dem Markt" sind keine zulässigen Nachweise im Sinne der EU 2017/745.
Neben den Anforderungen ans Produkt müssen in der technischen Dokumentation einige Mindestinhalte bereitgestellt werden. Dafür beschreiben die Anhänge II und III der MDR, was zu erstellen ist. Dabei ist darauf zu achten, dass sich all diese Inhalte zwischen unterschiedlichen Produkten und Herstellern massiv unterscheiden.
Da wir immer wieder gefragt werden:
Andere wichtige Bestandteile der technischen Dokumentation sind detaillierte Informationen über die Herstellung. Die Herstellung muss wirklich nachvollziehbar sein und es muss erkennbar sein, wo die Herstellung stattfindet. Dazu gehören auch alle Details zu den Qualitätskontrollen bzw. In-Prozess-Kontrollen und einer Chargenfreigabe.
Zu jedem Rohstoff / Komponente / Verpackung muss der jeweilige Lieferant ersichtlich sein. Sie benötigen also eine vollständig nachvollziehbare, dokumentiere Kette vom Rohmaterial zum wirklichen Medizinprodukt. Alle Materialien und alle beteiligten Stellen sowie Arbeitsschritte müssen nachvollziehbar sein.
Die folgende Grafik zeigt beispielhaft die Gliederung einer technischen Dokumentation nach Vorgabe der benannten Stelle DQS. Durch Anklicken des Bildes öffnet sich das hinterlegte PDF und Sie bekommen Einblick in die jeweiligen Unterordner der technischen Dokumentation.
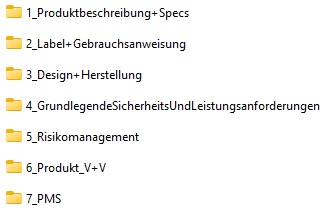
Hier können Sie die Vorlage kostenfrei herunterladen.
Selbstverständlich können die nicht anwendbaren Kapitel für Ihr Medizinprodukt Klasse I aus der Struktur entfernt werden. Dennoch ist es unserer Meinung nach ein gutes, weil vollständiges Beispiel einer technischen Dokumentation für unterschiedlichste Produkte.
Für alle, die sich jetzt Sorgen machen: Wir kennen uns damit gut aus und helfen gern dabei, die notwendigen Nachweise zu erstellen.
In der EU-Konformitätserklärung bestätigen Sie als Hersteller, dass das Produkt die Anforderungen der MDR einhält. Das ist im Grunde die Zulassung als Medizinprodukt der Klasse 1. SIe benötigen dabei keine Zertifizierungsstelle oder Behörde. Dazu muss man aber sagen, dass die Ausstellung nicht legal ist, wenn das Produkt die Anforderungen nicht erfüllt. Die Aufsichtsbehörden überprüfen regelmäßig Hersteller, die sich neu in DMIDS bzw. EUDAMED anmelden (später mehr dazu). Es ist also empfehlenswert, die Konformitätserklärung nur auszustellen, wenn die technische Dokumentation für das Produkt vorliegt und Sie ein QM-System haben.
Die EU-Konformitätserklärung muss die in Anhang IV der MDR genannten Punkte enthalten. Das folgende Bild zeigt den Ausschnitt der MDR aus Anhang IV.

Die SRN ist die Single Registration Number, die Sie bei der Registrierung in EUDAMED erhalten. Stand heute (22.12.2022) ist die Verwendung von EUDAMED noch freiwillig, weil die Datenbank nicht voll funktionsfähig ist. Es kann also sein, dass Sie die EU-DoC (EU-Konformitätserklärung) eine ganze Weile ohne SRN ausstellen können.
Die Basis-UDI-DI ist eine eindeutige Nummer, über die eine Zuordnung von Produkt und Ihnen als Hersteller möglich ist. Diese ist nicht mit der UDI-DI (z.B.GTIN) und der UDI-DI (z.B. Chargennummer + Haltbarkeitsdatum) zu verwechseln.
DMIDS ist das in Deutschland aktuell verwendete System, um sich als Hersteller von Medizinprodukten bei den lokalen Behörden anzumelden. Dort werden auch die Medizinprodukte angemeldet, die Sie als Hersteller vertreiben. Die Verwendung von DMIDS ist aktuell gesetzlich notwendig.
Es gibt eine weitere Datenbank bzw. System auf europäischer Ebene namens EUDAMED. EUDAMED ist aktuell aber noch nicht voll funktionsfähig und daher ist es gesetzlich noch nicht gefordert, Produkte bei EUDAMED anzumelden. Die Anmeldung von Produkten in EUDAMED ist derzeit also freiwillig.
In Ihrem QM-System sollte detailliert beschrieben sein, wie die technische Dokumentation (TD) dauerhaft aufrechterhalten werden kann. Denn die TD wird nicht bloß einmal initial erstellt und gilt dann bis in alle Ewigkeit.
Es ist gesetzlich notwendig, eine sogenannte Überwachung nach dem Inverkehrbringen durchzuführen. Mehr dazu hier. Dabei wird vom Hersteller überprüft, ob das eigene Produkt sich im Markt so verhält, wie es sein sollte. Es wird also geprüft, ob das Produkt im Markt irgendwelche Probleme hinsichtlich Sicherheit oder Leistung macht. Die daraus gewonnenen Erkenntnisse werden in der technischen Dokumentation abgelegt.
Auch das Risikomanagement ist ständig zu aktualisieren. Sie bekommen beispielsweise eine Reklamation in der ein Risiko oder Produktproblem bekannt wird, das noch nicht in der Risikoanalyse betrachtet wurde. Dann müssen Sie dies nachholen und die Risikomanagement-Akte aktualisieren.
Auch jede Änderung am Produkt-Design, den Etiketten, der Gebrauchsanweisung - alles muss in der technischen Dokumentation aktualisiert werden, damit diese stets dem aktuellen Stand entspricht.
Lokale Aufsichtsbehörde & Inspektionen - Hilfe! Was jetzt?
Hat sich Ihre lokale Aufsichtsbehörde zu einer Inspektion angemeldet und Sie haben Sorge, dass Ihre technische Dokumentation / QM-System nicht ausreichen?
Hat Ihre lokale Aufsichtsbehörde bereits Abweichungen formuliert und eine Frist gesetzt, in der Sie diese abstellen müssen?
Bitte kontaktieren Sie uns zügig, damit eine fristgerechte Reaktion möglich ist. Gemeinsam finden wir eine Lösung!
Streitfall Klassifizierung: Mein Produkt ist Klasse 1 - die lokale Behörde sieht das anders.
In diesem Fall kann es sein, dass eine Entscheidung durch das BfArM nötig wird. Das BfArM schreibt dazu folgendes auf seiner Website:
"Für eine Abgrenzungs- bzw. Klassifizierungsentscheidung durch das BfArM muss dem BfArM ein formloser Antrag auf Klassifizierung und/oder Abgrenzung gemäß § 6 Abs. 2 MPDG vorgelegt werden. Grundsätzlich antragsberechtigt sind eine deutsche Benannte Stelle, der Hersteller oder die zuständige Landesbehörde."
Erfahrungsgemäß gibt es insbesondere bei Medizinprodukten der Klasse 1 Unstimmigkeiten bezüglich der Klassifizierung, wenn es sich um Software handelt.
Es herrscht eine weit verbreitete Meinung, dass Software als Klasse 1 unter MDR quasi gar nicht möglich ist. Das stimmt so allerdings nicht. Regel 11 der MDR beschreibt klar, wann eine Software in Klasse 1 fallen kann. Die Regel beschreibt auch klar, was eine Software nicht tun darf, wenn man eine Software in Klasse 1 einstufen möchte.
Zugegeben, die Möglichkeiten einer solchen Software in Klasse 1 sind beschränkt. Aber es gibt sie. Und das ist auch gut so. Leider sehen einige lokale Behörden das anders - Aber in diesen Fällen empfiehlt es sich, für Rechtssicherheit mittels BfArM Antrag zu sorgen.